





间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)是非小细胞肺癌(non-small cell lung cancer,NSCLC)中常见的致癌驱动基因, 发生率约为5%,常见于无吸烟史、女性和年轻(中位确诊年龄为51岁)患者中[1-2]。目前,第三代ALK酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKI)(包括克唑替尼、塞瑞替尼、阿来替尼、布加替尼、恩沙替尼和劳拉替尼)在ALK融合基因阳性的NSCLC患者中均取得了优异的治疗效果。近年来的研究数据表明,TKI序贯治疗能极大程度地延长患者的生存期,ALK基因突变也因此有“钻石突变”美誉[3-4]。尽管靶向治疗疗效显著,患者最终会对靶向治疗产生获得性耐药[5]。ALK融合基因阳性NSCLC患者的耐药机制包括依赖ALK信号转导通路(ALK基因扩增和激酶域突变)、不依赖ALK信号转导通路(旁路激活和组织类型转化)以及伴随基因(EGFR、KRAS和TP53等)改变。本文聚焦于ALK耐药突变谱探索后续治疗方案和应对策略。
1
ALK基因背景与耐药
ALK基因位于染色体2p23位点,正常状态下可编码受体酪氨酸激酶(receptor tyrosine kinase,RTK)。RTK包含三部分结构,即细胞外区(接受外界信号)、跨膜区及位于细胞内的功能结构域。ALK基因主要在胚胎时期表达(合成蛋白质),促进神经系统的发育,之后进入休眠状态[6]。ALK基因变异(包括融合突变、点突变和基因扩增)导致激酶域异常激活,出现病理性ALK信号,诱发体细胞增殖和凋亡抑制,常出现在间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL)、炎症性肌成纤维细胞瘤(inflammatory myofibroblastic tumor,IMT)及NSCLC等恶性肿瘤[7-9]。在NSCLC中,ALK融合突变是最常见的变异类型,以棘皮动物微管相关蛋白样因子4(echinoderm microtubule associated protein like 4,EML4)-ALK融合为主,约占85%,其他伴侣分子包括KIF5B、HIP1、GCC2及PLB1等[10-13]。
ALK点突变和基因扩增是导致NSCLC对ALK-TKI耐药的重要机制。所有接受TKI治疗的患者在耐药后都能发现ALK基因扩增[14-15]。点突变在初治患者中并不常见[16],绝大多数都在癌细胞对TKI耐药的情况下出现[14]。ALK基因的常见突变位点于RTK细胞内激酶结构域,突变后导致相应部位氨基酸改变,蛋白质构象变化,形成空间位阻,干扰TKI与靶点的结合,导致获得性耐药[14,17]。
由于肿瘤发展的异质性,ALK基因突变位点和类型丰富而复杂[14,18-19]。首先,第一/二代TKI治疗失败后ALK基因的耐药变异以单点突变为主。约20%的患者在接受第一代TKI治疗进展后出现耐药突变,以L1196M、G1269A、C1156Y和F1174L为主;第二代TKI耐药后点突变的发生率更高(>50%),类型更丰富,例如G1202R/del、F1174C/V和I1171T/N/S等[14,18, 20]。其次,第二代TKI耐药后ALK双重突变和“脱靶”比例显著增加[14,21]。第三代TKI耐药后几乎均为复合突变,并且耐药程度更高,如G1202R+L1196M[22]。多代ALK-TKI治疗进展后,原有耐药位点发生变化,野生型的比列升高,耐药机制可能更为复杂[18]。
1.1 变异体与耐药突变
EML4-ALK变异体是影响获得性耐药的重要因素。已有研究[23-24]发现,V3变异体发生继发性耐药突变的概率高于V1、V2,突变类型及复合程度相对耐药性更高、更独特,如在V3中常见的ALK突变是C1156Y、F1174C/V、G1269A、I1171T、L1152R/V和G1202R,而L1196M突变在V1变异体中更常见。尤其, G1202R在V3变异体的发生率较V1变异体高( 32% vs 0%,P = 0.001)[23],对G1/G2 ALK-TKI呈现普遍的高度耐药性,只有劳拉替尼对其效果明显[18,22]。
1.2 共存突变与耐药突变
在ALK融合基因阳性的NSCLC患者中,最常见的伴随突变是肿瘤蛋白P53(tumor protein P53,TP53)基因突变(>20%)[25-27]。基线时具有TP53突变(TP53-mutant,TP53-MUT)患者的中位总生存期(median overall survival,mOS)显著劣于TP53野生型肿瘤(TP53-wild type,TP53-WT)的患者(44个月 vs 62个月,P = 0.018)[28]。TP53共突变患者的突变负荷(tumor mutation burden,TMB)明显高于野生型患者(7.07±1.25 vs 3.20±0.33,P = 0.003),且耐药突变的发生率也高于TP53野生型患者,表明TP53突变可能导致其肿瘤抑制功能受损,影响基因组稳定性,促进基因进化,最终诱发耐药突变[29]。
2
ALK-TKI获得性ALK激酶域耐药机制和治疗策略
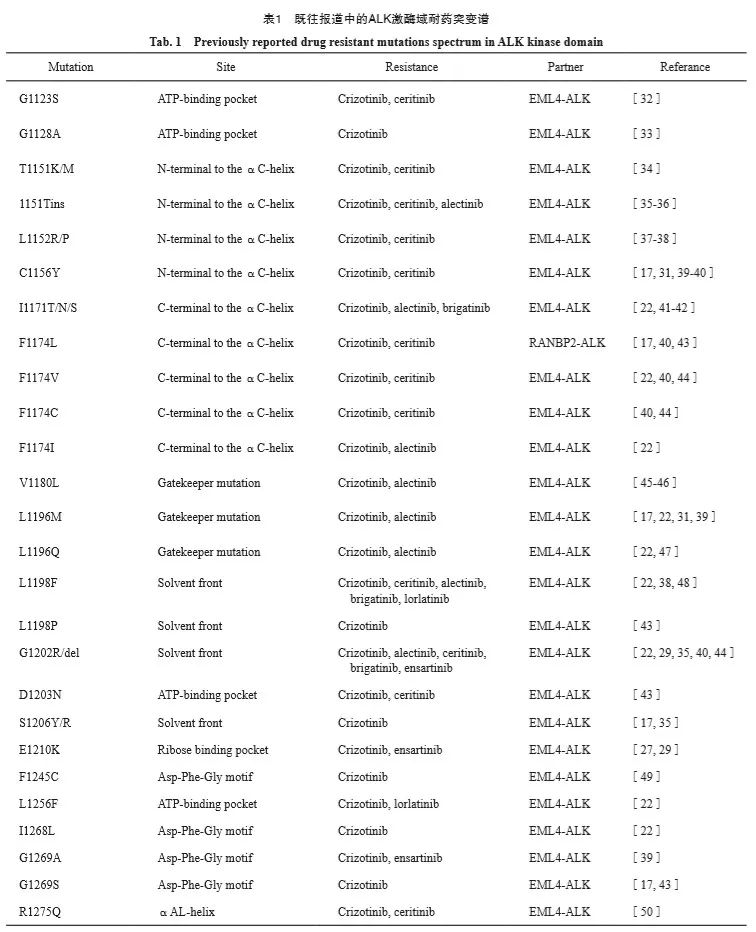
ALK基因激酶结构域由较小的N瓣和较大的C瓣构成,通过“铰链区”链接[30]。ATP结合位点位于N瓣和C瓣之间,核苷酸定位环(P环)的下方。ALK-TKI通过与 ALK激酶结构域的ATP结合位点竞争性结合来达到抗肿瘤疗效[30]。ALK激酶域突变可分为5个热点区域:① ATP结合域(ATP-binding pocket);② 核糖体结合口袋区域(ribose binding pocket);③ α-C螺旋的N段或C段(N/C-terminal to the αC-helix);④ 激酶铰链区(solvent front);⑤ DFG基序(Asp-Phe- Gly)[17]。常见的具有较强耐药性突变位点是“门控”的L1196、溶剂前沿的G1202和L1198以及DFG 基序附近的G1269等[31]。
2.1 克唑替尼耐药谱
研究[14-15]发现,克唑替尼(crizotinib)治 疗失败后,30%~45%的患者经克唑替尼治疗后会发生ALK激酶区基因突变和基因扩增。2010年,Chio等[31]首次发现L1196M、C1156Y两种突变,并且确认是克唑替尼的耐药机制。ALK-1196基因位于ATP结合域的底部,属“门控”关键位置,突变后形成空间位阻,影响药物与靶点结合,是克唑替尼治疗出现进展后常见的耐药突变之一[31]。体外实验通过构建克唑替尼耐药细胞系时发现,耐药细胞不仅有获得性L1196M突变,还有EML4-ALK基因的拷贝数增加,提示获得性耐药可能是个动态过程,涉及基因扩增和点突变[15]。随着NGS研究不断深入,ALK-TKI的耐药突变谱逐渐清晰,L1152R/P、F1174L/V、G1269S、D1203N、I1171T、S1206Y及1151Tins等耐药性突变被陆续发现(表1)。
2.2 第二代ALK-TKI
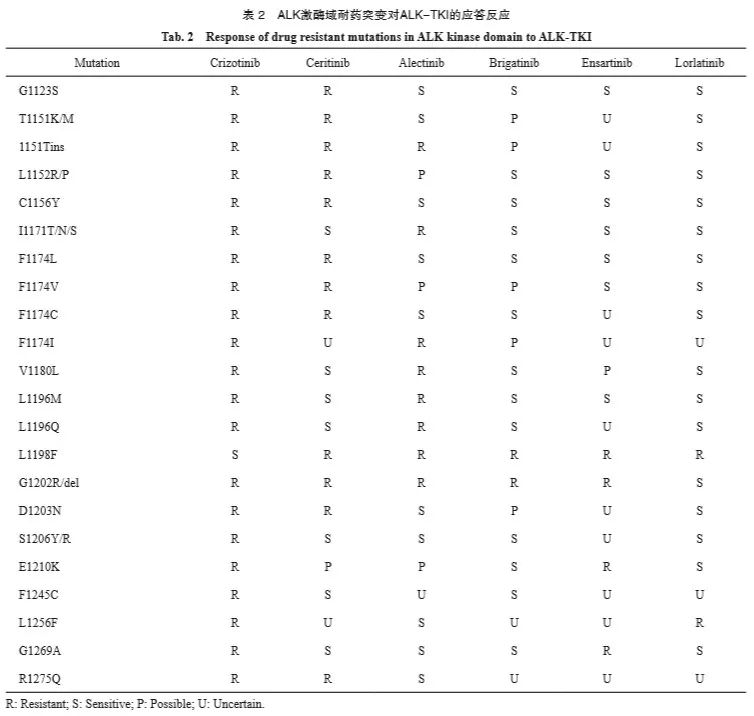
在克唑替尼治疗失败后,超过70%的患者选择接受后代ALK-TKI进行治疗,继续延长生存期[5]。相对于克唑替尼,第二代ALK-TKI(塞瑞替尼、阿来替尼、布加替尼和恩沙替尼)对ALK激酶具有更低的半数最大抑制浓度(median inhibition concentration,IC50),可覆盖大部分的ALK耐药突变(表2),并显示出更好的中枢 神经系统(central nervous system,CNS)渗透性[25-40](表2)。
塞瑞替尼(ceritinib)与ALK的结合位置是由铰链区、P环、α-C螺旋和激活环组成的ATP结合口袋,主要限制野生型和突变型复合物中P环的构象改变[30]。在ASCEND-4研究[51]中, 一线塞瑞替尼的中位无进展生存期( median progression-free survival,mPFS)显著优于化疗(16.6个月 vs 10.6个月,P<0.001);在没有基线CNS转移的患者中,塞瑞替尼的mPFS长达26.3个月。对比化疗,克唑替尼进展和1/2 线化疗失败的患者仍然能从塞瑞替尼治疗中获益(客观缓解率,overall response rate,ORR 42.6% vs 6.0%),mPFS(5.4个月 vs 1.7个月)更长[52]。在临床前模型中,塞瑞替尼能有效地抑制克唑替尼常见的耐药性突变,如L1196M、G1269A、I1171T及S1206Y等[30,40]。而且L1196M和I1171T/N/S突变点是塞瑞替尼独特的治疗位点[41],ALK F1245C突变也对塞瑞替尼非常敏感[49]。而C1156Y、1151Tins、L1152R/P、R1275Q、G1202R/del、F1174V/C/L、T1151K及G1123S等突变会影响塞瑞替尼的结合位点产生耐药[14,32,34,40,50]。其中,G1202R和F1174C/L是塞瑞替尼治疗失败后的主要耐药突变位点[14,53]。G1123S 、T1151K和F1174C突变可导致P环出现波动,影响塞瑞替尼和P环之间较强的相互作用,导致耐药性的产生[32,34]。 而D1203、C1156、R1275、L1198及L1152等突变位点主要是由于影响塞瑞替尼与ALK的结合环境,干扰靶点的结合从而导致耐药性。ALK T1151Sins突变导致患者对塞瑞替尼、阿来替尼产生耐药性,对劳拉替尼敏感[36]。
阿来替尼(alectinib)是经优化溶剂相互作用以及调节ATP结合位点后的高效选择性TKI,与野生型ALK结合位点主要在铰链区、P环及附近结构[54]。在ALEX研究中,阿来替尼组的mPFS为34.8个月,显著优于克唑替尼组的10.9 个月(HR = 0.43,P<0.000 1),并且显著改善ORR [55]。阿来替尼目前已成为ALK+晚期NSCLC患者的一线治疗优先选择。对于克唑替尼难治性患者,是否携带ALK耐药突变基因影响阿来替尼的治疗效果,不携带耐药突变的患者的mPFS(9.1个月 vs 5.6个月)和ORR(45% vs 35%)更好[46]。在体外实验中,阿来替尼对大部分常见耐药突变有较高活性,如C1156Y、F1174L、G1269A及D1203N等[22,50]。而研究[14,46]发现,阿来替尼治疗失败后最常见的突变是G1202R和I1171N/S/T。ALK G1202R/del/ K突变会导致阿来替尼耐药性[22,56-57]。由于L1198、V1180、I1171N和L1196位点突变会影响ATP结合入口和铰链区的构象变化,诱导阿来替尼与ALK之间的“锁扣”作用发生变化[48]。诸如两种“门控”突变V1180L、ALK L1196M/Q及I1171T/N/S突变,对阿来替尼和克唑替尼均会产生耐药,但对塞瑞替尼、布加替尼及劳拉替尼等后代抑制剂敏感[22,42,45-47,58]。
布加替尼(brigatinib)是ALK/EGFR双重抑制剂,对ALK L1196M突变和EGFR T790M突变高度敏感[59]。ALTA-1L研究[60]显示,布加替尼的mPFS优于克唑替尼(24.0个月 vs 11.0个月, P <0.001)。在克唑替尼耐药后患者中,布加替尼的mPFS为16.7个月[61]。在塞瑞替尼、阿来替尼甚至经历多线治疗(至少2种ALK-TKI)中, 布加替尼仍然能获得7个月的mPFS[62-64],从而确立了布加替尼在一代或其他二代ALK-TKI进展后的治疗地位。在临床前模型中,布加替尼对于ALK+和ALK-细胞均有更强的抑制活性,对于其耐药突变谱均有更高的抑制效力,对G1202R突变有中度抑制能力[38]。尽管对G1202R有抑制活性,高达70%的患者在接受布加替尼治疗失败后检测到新发的G1202R突变[14]。临床研究显示,布加替尼确实对许多继发性ALK突变显示出抗肿瘤活性,包括L1196M、F1174L/V、G1269A、I1171N、L1198F和 V1180L等,与临床前试验显示的广谱活性相似[64]。布加替尼主要通过氢键与ALK-K1150、L1196、L1198和E1210残基产生相互作用,能耐受ATP结合口袋中的任何单一突变,但G1202R除外[65]。由于G1202R突变的存在,导致患者对布加替尼和神经营养性酪氨酸激酶(neurotrophic tyrosine kinase,NTRK)抑制剂的反应较差。因此,G1202R突变可能会导致布加替尼原发性耐药[66]。此外,有研究[14]显示,S1206C也会导致布加替尼耐药。
恩沙替尼(ensartinib)已获批用于治疗克唑替尼治疗后进展的或不耐受的ALK阳性的局部晚期或转移性NSCLC患者。在TKI初治患者中,恩沙替尼的mPFS(25.8个月 vs 12.7 个月;P<0.001)和12 个月CNS进展率(23.9% vs 4.2%;P = 0.001)均优于克唑替尼[67]。在克唑替尼难治性患者中,恩沙替尼的mPFS为9.6个月,颅内ORR可达70%[68]。无论有无ALK激酶域耐药突变,恩沙替尼都表现出相似的ORR,对F1174L/V、C1156Y、T1151、G1123S和L1198F突变敏感性较高,再进展患者中最常见的继发性耐药突变是G1269A、G1202R和E1210K,而G1269A对其他第二代抑制剂敏感,G1202R对劳拉替尼的治疗效果最明确[27,29,68]。
2.3 第三代ALK-TKI
劳拉替尼(lorlatinib)是第三代ALK-TKI, 拥有更高的CNS渗透性,且可更广泛有效地克服已知的耐药性突变(表2)。CROWN研究[69]显示,劳拉替尼12个月无进展生存率(78% vs 39%) 和ORR(76% vs 58%)显著优于克唑替尼。在克唑替尼耐药的患者中,劳拉替尼的ORR为73%(95% CI:60%~84%),mPFS为11.1个月(95% CI:8.2 个月~未达到),而且无论是否存在可检测的ALK突变,患者的ORR是相似的[18]。在二代ALK-TKI耐药背景下,劳拉替尼的抗肿瘤反应在不同ALK基因激酶域位点突变状态下产生差异,基于组织检测的ALK基因突变阳性患者的ORR(69% vs 27%)和PFS(11.0个月 vs 5.4 个月) 明显优于突变阴性患者[18]。因此,在曾接受过至少一种ALK-TKI的患者中,对肿瘤组织进行再次检测是很有必要的,ALK基因突变情况可能提示患者接受劳拉替尼的疗效及持久获益程度。
劳拉替尼对几乎所有已发现的耐药性突变有良好治疗效果,包括已知的高耐药性突变1151Tins、G1202R、I1171N和F1174L等(表2)。特别是对ALK-TKI普遍耐药的G1202R/del,劳拉替尼显示明显的抑制效果,ORR可达到57%,mPFS为8.2个月[18,22]。而ALK L1256F会导致劳拉替尼耐药,但该位点对阿来替尼敏感性很高。此外,劳拉替尼耐药后发生的L1198F突变会重新对克唑替尼敏感[22]。既往研究[19]还 发现,劳拉替尼进展后更容易出现双重或多重复合突变,某些复合突变对第一代/第二代TKI会重新敏感,如V1185L+L1196M复合突变,然而很多高度耐药性的复合突变依然得不到有效抑制,如G1202R+L1196M,这种复合突变是目前劳拉替尼程度最高的耐药突变(IC50 = 1 000 nmol/L),对所有的ALK-TKI都具有抗性[19]。
3
Solvent-front区域突变及治疗对策
Solvent-front区域是ALK激酶域的溶剂前沿,由突变诱导的构象变化和ALK-TKI与结合口袋之间的快速解离过程导致耐药性[54]。第二代TKI治疗失败后的耐药突变更容易出现于Solvent-front区域,比如G1202R/del、D1203N及L1198F[18,27]。ALK L1198F突变导致蛋白ATP结合位点的构象变化,导致ALK激酶域结合位点与克唑替尼和劳拉替尼之间的亲和力的变化,使得癌细胞对劳拉替尼耐药,而对克唑替尼再次敏感[70]。顽固性的溶剂前沿G1202R突变对一代/二代TKI保持普遍高水平耐药,是二代TKI治疗失败后最常见的耐药突变。这与克唑替尼的结合位点毗邻Arg-1202,塞瑞替尼、阿来替尼 的结合位点位于铰链区相关[14,53]。除了劳拉替尼能有效抑制[18,22],新型抑制剂ZX-29也能克服G1202R突变导致的耐药性[71]。针对溶剂前沿区域(G1202R)合成的嘧啶-2,4-二胺衍生物对部分耐药突变有抑制活性,“半游离尿素” 基团(仅有1个-NH部分)对最常见的L1196M 突变(IC50 = 0.91 nmol/L)以及G1202R(IC50 =4.3 nmol/L)有显著的抗肿瘤效果[72]。优化“尾巴”吡啶酮基序列,得到的2-氨基吡啶衍生物对L1196M(IC50 = 45 nmol/L)、G1202R(IC50 = 22 nmol/L)和ROS1(2.3 nmol/L)也表现出令人满意的抗肿瘤活性[73]。这些研究表明某些ALK特定区域的化学衍生物将是克服临床ALK激酶域耐药突变的有效方法。
4
复合耐药突变和治疗对策
ALK-TKI的多次序贯治疗后出现的双重复合突变已逐渐被认知。第二代ALK-TKI耐药后已出现一定比例的双重突变。V1185L+L1196M 双重突变对阿来替尼反应较差,但是V1185L、L1196M和复合突变均对塞瑞替尼敏感[22]。I1171T+E1210K复合突变在塞瑞替尼耐药后出现[21]。E1210K+D1203N和F1174C+D1203N复合突变赋予克唑替尼和第二代抑制剂耐药性,劳拉替尼可克服[14,22]。而劳拉替尼治疗后更容易出现多重复合突变[19,22]。
研究[19]显示,接受连续靶向治疗后,有35%的患者在劳拉替尼治疗失败后出现ALK复合突变。由于ALK G1202R突变是第二代ALK抑制剂耐药后最常见的激酶结构域突变,含G1202R的复合突变可能成为接受序贯第二代和第三代TKI后再次进展的患者中最常见的靶向耐药机制[19]。Okada等[22]在劳拉替尼治疗失败后的患者中发现,存在I1171N- 双重突变(I1171N = L1198F、+L1196M、+T1151K、+C1156Y、+F1174I、+L1198H、+L1256F和+G1269A)及G1202R-双重突变(G1202R = L1198F、+G1269A、+L1196M、+F1174C、+F1174L和S1206Y)。此外,还有I1171S+G1269A、C1156Y+G1269A和高水平耐药的L1196M+D1203N等复合耐药突变[20,74-75]。
G1202R+G1269A对劳拉替尼、塞瑞替尼耐药,虽然对布加替尼中度敏感,但是由于G1202R顽固耐药,临床应用布加替尼的效果可能受限[14,22]。布加替尼对I1171N复合突变( +L1198F、+L1196M、+L1256F和+G1269A )有抑制效果;塞瑞替尼可抑制I1171N+L1196M和+G1269A;阿来替尼可抑制I1171N+L1256F[22]。值得注意的是,由于克唑替尼对L1198F的敏感性较高[59],对劳拉替尼耐 药的L1198F突变可再次从克唑替尼治疗中获益,也能克服L1198F-复合突变,如I1171N/G1202R/ C1156Y+L1198F突变[19,22]。其他复合突变类型,如I1171S+G1269A、G1202R+S1206Y、L1196M+D1203N和C1156Y+G1269A,再次应用第二代TKI可能对肿瘤有抑制效果,如塞瑞替尼、布加替尼[20,74-75]。此外,第三代FMS样酪氨酸激酶-3(FMS-like tyrosine kinase-3,FLT3)抑制剂吉尔替尼(gilteritinib)对于I1171T/N/S单突变及I1171N-双重突变(+L1198F、+L1256F、+L1196M及+F1174I)有显著的效果[76]。
第四代TKI(TPX-0131和NVL-655)表现出显著的“双突变活性” ,并且具有更强的CNS渗透性和对野生型ALK的抑制能力[77-78]。 除了对已知单发耐药突变的广谱活性TPX-0131对L1198F+G1202R、L1198F+L1196M、L1198F+C1156Y、E 1210K+S1206C 、T1151I+L1152ins 和G1202R+L1196M 有抑制能力。 然而,G120 2R+G1269A和G1202R+G1269A+L1204V复合突变仍会导致TPX-0131耐药[78]。目前第四代TKI已进入临床试验阶段,有望突破复合耐药突变的瓶颈。
5
展望
目前,第三代ALK-TKI在ALK融合基因阳性的NSCLC患者中均取得了显著的治疗效果,然而,耐药所致靶向治疗失败不可避免。ALK激酶域突变是ALK-TKI的重要耐药机制之一,认识和探索ALK激酶域耐药突变的发生机制和优化未来应对策略是ALK+NSCLC治疗中亟待解决的问题。迄今为止,ALK激酶域耐药突变大多为是临床前试验、小样本检测和个案报道,缺乏大规模临床研究数据明确TKI治疗后耐药突变的发生机 制和对后代TKI的应答反应。目前,在克唑替尼进展后有第二代或第三代TKI可供选择,而第二代TKI治疗出现疾病进展后,仅有劳拉替尼作为“最后手段”。新一代TPX-0131和NVL-655在临床前研究中以表现出优异的抑瘤效果,尤其是能够克服ALK复合耐药突变,但仍需要临床试验的验证。同时,考虑到肿瘤的时空异质性以及个体耐药机制差异,仍建议在ALK-TKI治疗出现疾病进展后再次检测(包括活检和基因检测), 有助于识别难治性和重敏性ALK基因激酶域突变,明确TKI的耐药机制,开展更具针对性的基础和临床转化研究,确立更加精准有效的治疗策略。
利益冲突声明:所有作者均声明不存在利益冲突。
[参考文献]
声明:本文来源中国癌症杂志,仅为交流学习。内容仅代表作者个人观点,望大家理性判断及应








