准确检测体细胞突变并进一步解析癌症中的基因组不稳定性对于精准癌症治疗至关重要。近年来,许多基因组学研究极大地提高了人们对癌症基因组不稳定性和一般癌症生物学的理解。SEQC-2联盟更是建立了配对的肿瘤-正常参考样本和参考调用集,用于使用不同的测序平台和生物信息学分析方法对体细胞突变检测进行基准测试。这些研究为评估癌症诊断中体细胞突变检测的准确性和可重复性、设计个性化癌症免疫疗法以及分析可能干扰基于基因编辑疗法的潜在脱靶效应提供了重要资源。
目前,人们主要依赖于标准的人类参考基因组(如GRCh38)作为发现体细胞事件和定义高置信度参考体细胞调用集的基准。但GRCh38存在一些未解决的组装问题。因此,科研人员一直提倡使用个性化的单倍型特异性基因组组装。
DNA测序成本的大幅降低为生成高质量的单个基因组从头组装提供了绝佳的机会。但迄今为止还没有系统的研究使用个性化基因组作为体细胞突变检测的参考,特别是在配对的肿瘤-正常样本中。
近日,美国洛马林达大学和美国食品和药物管理局药物评估和研究中心的联合研究团队在Genome Biology上发表了题为“Personalized genome assembly for accurate cancer somatic mutation discovery using tumor-normal paired reference samples”的文章。研究团队结合多种测序技术生成了首个肿瘤-正常配对基因组的从头组装,包括核基因组和线粒体基因组,其样本来自一个三阴性乳腺癌患者。研究发现个性化参考基因组不仅改善了短读长和长读长测序数据的reads比对,还提高了体细胞SNV和SV的检测精度。

文章发表在Genome Biology上
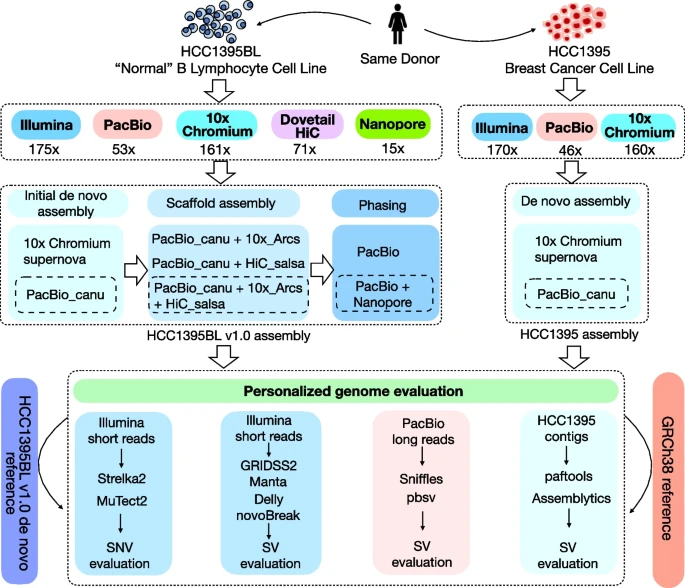
研究团队使用来自五个不同平台的测序数据组装正常参考样本(B淋巴细胞系HCC1395BL)的基因组,使用来自三个平台的数据组装肿瘤参考样本(来自同一个体的HCC1395乳腺癌细胞系)。通过来自多种测序技术的数据(包括Illumina短读长测序、10X Genomics链接测序、PacBio长读长测序等)构建了一个工作流程来生成一个从头组装的个人基因组,并称其为“HCC1395BL_v1.0”。研究团队使用这个组装的基因组和GRCh38作为参考,进行了reads映射和体细胞变异分析。
研究团队将NCBI RefSeq转录集(排除所有假基因和Y染色体基因)与HCC1395BL_v1.0进行了比对(图2)。结果显示,19,325个RefSeq蛋白编码基因中有19303个(99.27%)可以成功地映射到HCC1395BL_v1.0上,比对一致性至少为95%,比对覆盖率为50%;在10,061个RefSeq非蛋白编码基因中,99.88%可以成功映射到HCC1395BL_v1.0上,一致性最低为95%,覆盖率为50%。
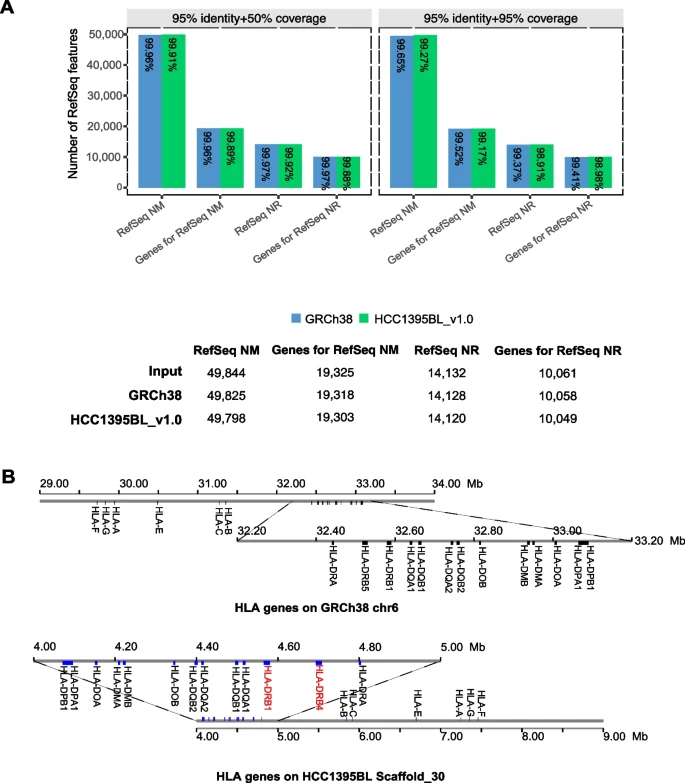
图2. HCC1395BL_v1.0和GRCh38上的RefSeq基因/转录本映射的总结,截断区为95%识别率+ 50%覆盖率与95%识别率+ 95%覆盖率。
接下来,研究团队将HCC1395BL_v1.0和GRCh38作为reads映射的参考并对其进行了比较(图3)。虽然在6个测序中心的12个WGS重复中,HCC1395BL和HCC1395细胞系的短reads与GRCh38原始组装(不包括替代位点支架除外)、HCC1395BL_v1.0的映射率非常相似,但与GRCh38相比,在HCC1395BL_v1.0上这些重复有所改善。
值得注意的是,研究团队观察到当个人基因组HCC1395BL_v1.0被用作参考时,HCC1395BL的库插入尺寸标准偏差平均小2.76,HCC1395平均小2.83。此外,HCC1395BL_v1.0上的reads覆盖率的标准偏差比GRCh38小得多,表明reads在HCC1395BL_v1.0上放置得更均匀。
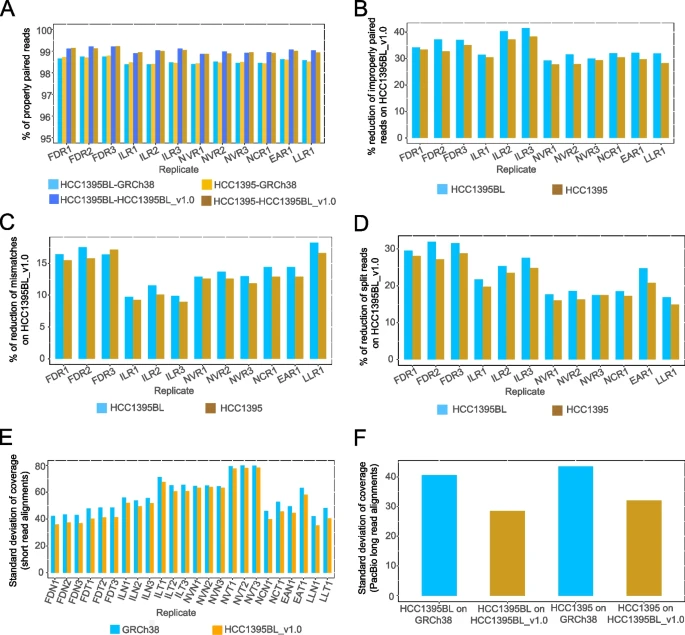
图3. 与GRCh38相比,个性化基因组HCC1395BL_v1.0参考的Illumina短读长和PacBio长读长映射的改进情况。
由于预测体细胞SV的基本算法不同,不同工具所报告的SV事件在数量、类型和大小方面可能存在很大差异。因此,研究团队选择了GRIDSS2/GRIPSS、Manta、Delly和novoBreak四种体细胞SV检测作为不同检测算法的代表,在HCC1395BL_v1.0、GRCh38分别作为参考时,评估了它们的相对性能(图4)。结果显示,在HCC1395BL_v1.0作为参考时检测到了更多的体细胞突变,无论是否包括易位变异检测(TRA)。
研究团队还观察到,GRIDSS2和Manta检测到的总SV计数高于Delly和novoBreak,表明与算法相对简单的检测方法相比,具有更复杂算法的检测方法可能更敏感。此外,Manta在HCC1395BL_v1.0上检测到20个插入变异,在GRCh38上检测到10个插入变异,表明个性化的HCC1395BL_v1.0作为参考时,对样本短读长测序数据的体细胞插入检测具有较好的敏感性。
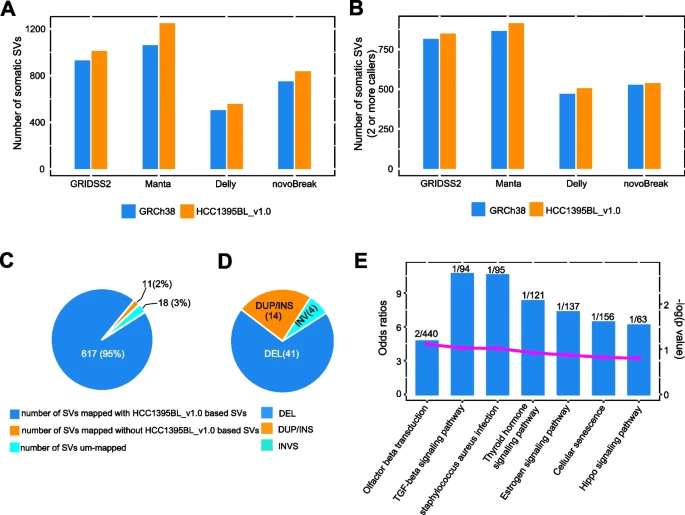
图4. 与GRCh38相比,使用HCC1395BL_v1.0参考的肿瘤-正常配对短读长WGS数据进行体细胞SV检测。
在扩展分析中,研究团队纳入来自6个测序中心的所有12个肿瘤-正常配对的WGS重复,以便分析与GRCh38相比,使用HCC1395BL_v1.0作为参考对4个检测方法的影响(图5)。结果显示,两个参考的倒置变异检测基本相似,只是在HCC1395BL_v1.0上多了1或2个倒置。对于DUP计数,GRIDSS2和Manta都报告在HCC1395BL_v1.0上多了10个(4.44%)和11个(4.54%),但Delly检测到的DUP计数没有变化。
通过将这些来自两个或多个重复的基于GRCh38的SV映射到HCC1395BL_v1.0参考上,发现GRIDSS2的24个SV、Manta的41个SV、Delly的43个SV和novoBreak的28个SV被认为是“未映射”或“在HCC1395BL_v1.0上已映射但没有匹配的SV”。当使用HCC1395BL_v1.0作为参考时,根据映射标准,GRIDSS2的61个SV、Manta的86个SV、Delly的61个SV和novoBreak的55个SV在HCC1395BL_v1.0参考上的相应位置缺乏基于GRCh38的SV。
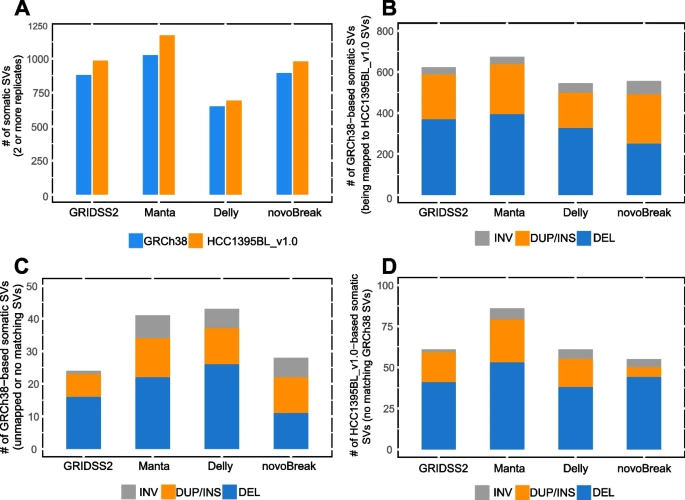
图5. 与GRCh38相比,HCC1395BL_v1.0上的4个短读调用器在两个或多个重复中检测到的体细胞SV。
综上所述,该研究证明个性化基因组不仅具有个人特异性的单倍型,还可以更好地表示样本中的基因组区域,包括临床相关基因。此外,个性化基因组能够更准确地检测成对肿瘤-正常样本中的体细胞突变,包括体细胞SNV和SV。特别是新的体细胞突变(SNV /SV)只在个性化基因组作为参考时被发现。
参考文献:
Xiao, C., Chen, Z., Chen, W. et al. Personalized genome assembly for accurate cancer somatic mutation discovery using tumor-normal paired reference samples. Genome Biol 23, 237 (2022). https://doi.org/10.1186/s13059-022-02803-x
声明:本文来源测序中国,仅为交流学习。内容仅代表作者个人观点,望大家理性判断及应用。